Tutorial - help section
Palantir utilities and functionalities
The screening and identification of secondary metaboism pathways and product by in silico analysis of organism genomes have been increasingly developed this last decade and currently plays an important supporting role in the discovery of new natural products. For contributing to this effort of providing tools for improving and accelerating the discovery of bioactive compounds, we devised Palantir with two categories of functionalities and utilities: (1) new functionalities for improving the annotation of modular NRPS (NonRibosomal Peptide Synthetases) and PKS (PolyKetide Synthetases) and enzymatic system, as well as, (2) springboard utilities for automating the parsing and querying of hundres/thousands of genome results.
Regarding NRPS/type 1 PKS functionalities, Palantir offers five functionalities for supporting their investigation on different aspects: (1) a delineation method of gene clusters in modules (set of domains responsible of the addition of a monomer to the synthesized product); (2) a "gap-filling" for checking domain gaps in gene clusters and filling these in case of protein signature matches. Moreover, (3) A dynamic elongation of the sequences core domains for optimizing the collected information of domain sequences (this method showed to improve the phylogenetic signal, for more in formation, see our [supplementary information](). We observed that NRPS and type 1 PKS gene clusters show a propensity for complex and versatile domain compositions and architectures. To help to overcome the undersanding of peculiar NRPS/PKS clusters, we provided in Palantir an "Exploratory" annotation mode of these consisting in a visualization of every domain signature matches without the constraints fo a pre-exising consensus of the cluster achitecture. Finally, an interactive visualization tool is provided for comparing antiSMASH, Palantir and "Exploratory" Palantir annotations. The pHMM database used for gap-filling and elongation mehtods contains the profiles used by antiSMASH authors since their version 3 (Weber et al., 2015), NRPS/PKS substrate predictor (Khayatt et al., 2013) and core fungal signatures from Bushley’s work (Bushley et al., 2010).
If analyzing and interpreting a few genome mining results is trivial thanks to GUI tools, such as antiSMASH, it requires a strong bioinformatics background to handle multidimensional data of thousands biosynthetic gene cluster (BGC) results, frequently obtained in large-scale genome mining projects. For supporting such projects, we provided several utilities in Palantir for handling large amount of BGC data: : (1) FASTA sequence extraction at any BGC level (cluster, gene, module or domain), (2) PDF/Word reporting and (3) relational (SQL) database generation for more advanced data analysis.
Palantir is available as a web tool and a fully documented (command-line) Perl package (https://metacpan.org/release/Bio-Palantir), the set of functionalities for Refining the NRPS and type 1 PKS annotation are available on these both supports. However, as methods for handling large amount of BGC data are intended to be applied to hundreds or thousands of antiSMASH reports, these latter are only available through the Perl package.
We hope Palantir to be useful in future small- and large-scale genome mining projects and natural products studies. For more information, we invite you to read our publication:
Browsing Palantir cyanobacterial database
The database provided on this website is based on the antiSMASH prediction of secondary metabolite gene clusters in 1489 cyanobacterial genomes (from a 1616 genome analysis, see Supplementary Information for the complete list, future link. For accessing the biosynthetic gene cluster (BGC) information, the data are organized following their taxonomy (lineage classification).
Also, for helping users investigating cyanobacterial secondary metabolites, we added the percentage of contamination next to the strain name (e.g., Richelia intracellularis UBA7409 [1.56%]). This assessment of the contamination level was performed by the pipeline described in Cornet et al., 2018.
Several options are available for browsing this database and are explained in the next sections.
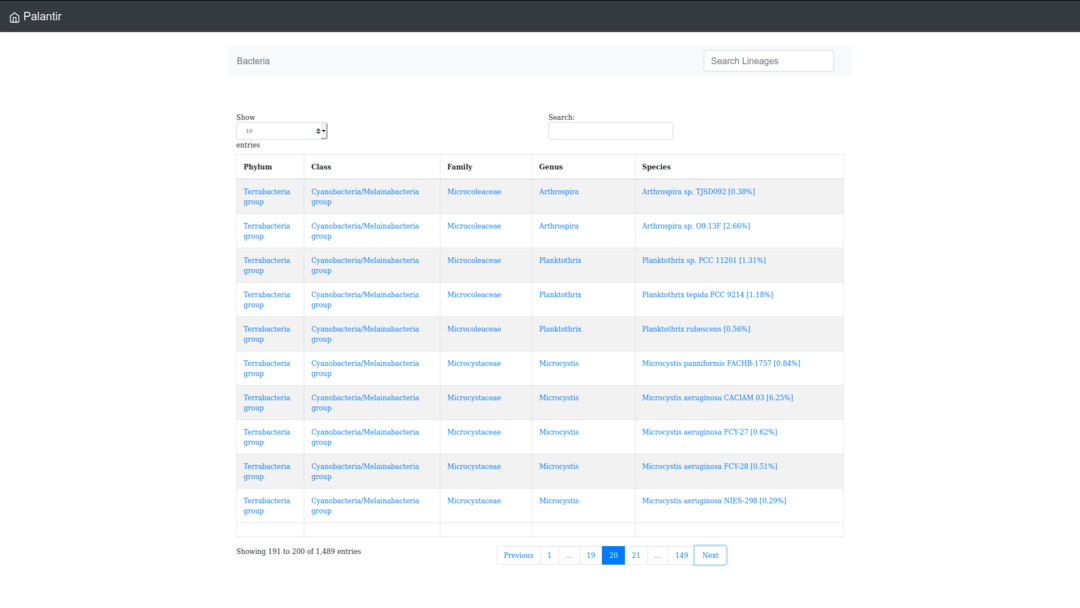
Browsing the cyanobacterial lineages by alphabetic order
By clicking on the "Browse the database" tab, you can access to the taxonomic information of cyanobacterial lineages and browse these by alphabetic order.
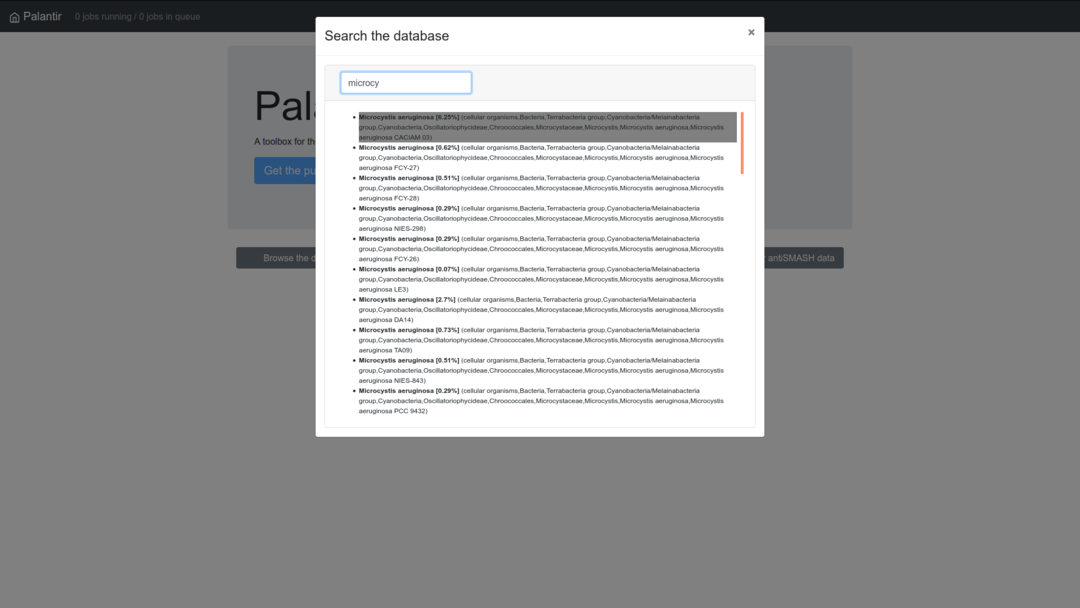
Searching a specific taxonomic level or genome accession number
The "Search lineages" tab allow you to directly look for a specific taxonomic level, such as genus or species names, or the accession number of a genome, such as GCA_003388675.1.
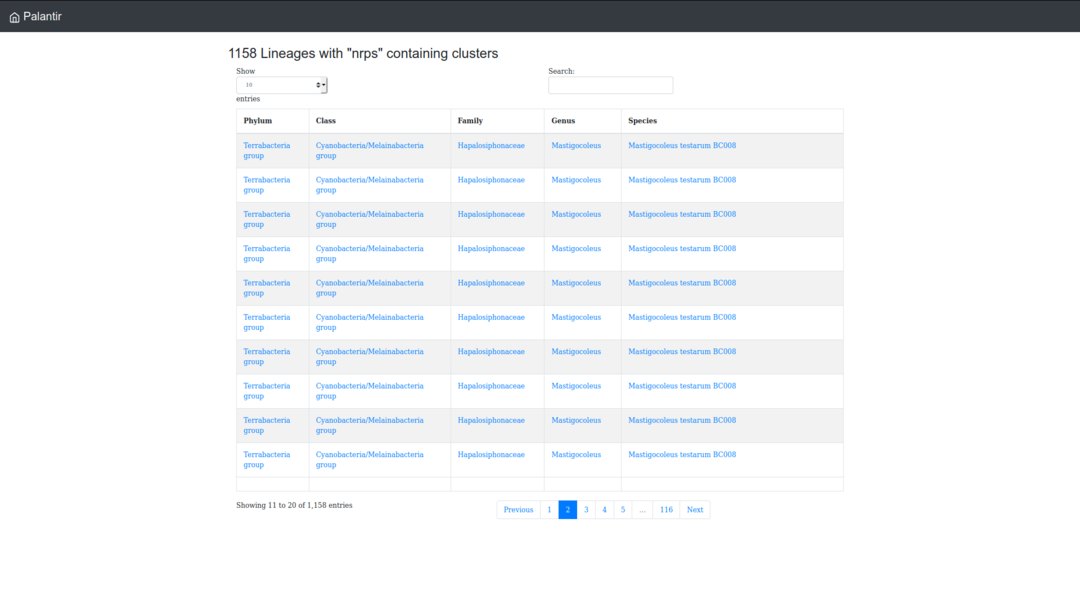
Filtering lineages that contain a specific gene cluster type
For filtering and isolating cyanobacterial lineages that contain a particular type of gene cluster, you can use "search by Cluster type" and select a gene cluster type (the names are extracted from antiSMASH reports, see antiSMASH explanation list, for more details).
Analyzing your own data
Palantir functionalities can be applied to your data by uploading antiSMASH report on our web interface. For this, after running your analysis on the antiSMASH website, you can access the repository of your analysis by clicking on the "Download" tab.
Then, after unzipping the folder, select either the biosynML.xml (antiSMASH versions 3 and 4) or the regions.js file (>= antiSMASH version 5) and upload it on Palantir website with the "analyze your antiSMASH data" tab. A label is also suggested to name and easier retrieve your analyses.
For trial, you can skip these steps by simply using the "Load test data" button.
Once the job submitted and running through our server, the results will be available after a few minutes. Your email address will optionnally be asked for helping you to access the results later, but you can also pin the link if it suits you more.
Interpreting Palantir results
When accessing results, you first arrive on an overview page summing up every biosynthetic gene cluster (detected by antiSMASH) accompanied by a representation of its genes.
\begin{figure}[h!] \centering \includegraphics[width=1\textwidth]{images/overview_1080x608.png} \caption{\small{traditional vs genetic view of protist diversity. Based on DelCampo \textit{et al}., 2014 data and modified from Maria Ferrer-Bonet adaptation.}} \end{figure}
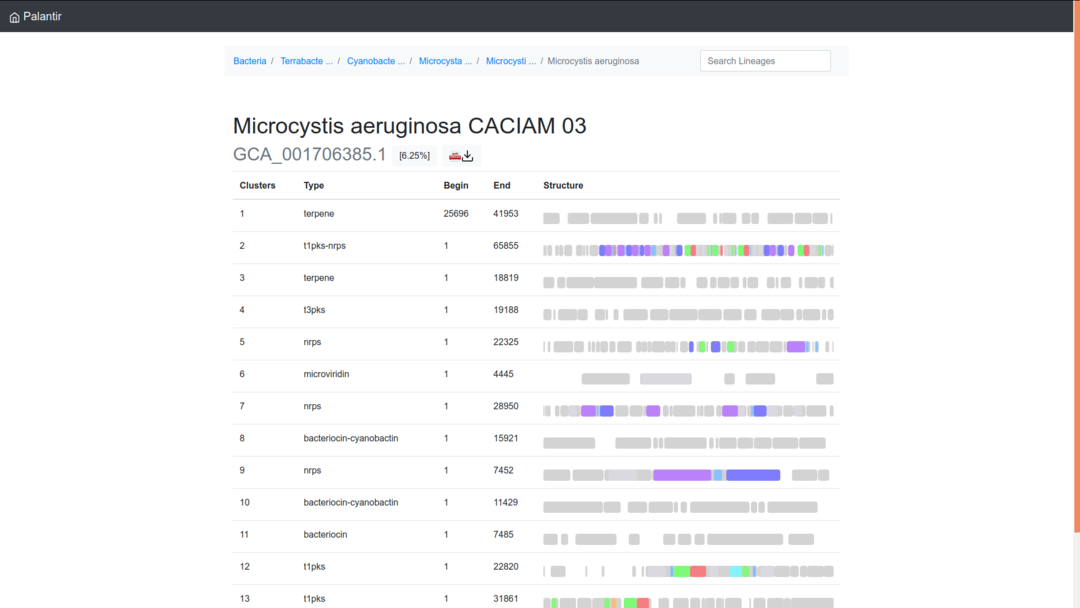
From this page, you can then choose a gene cluster based on its type, coordinates and genes illustration (connect this example here). For instance, you will obtain this view if you choose the second cluster (hybrid PKS type 1 - NRPS).
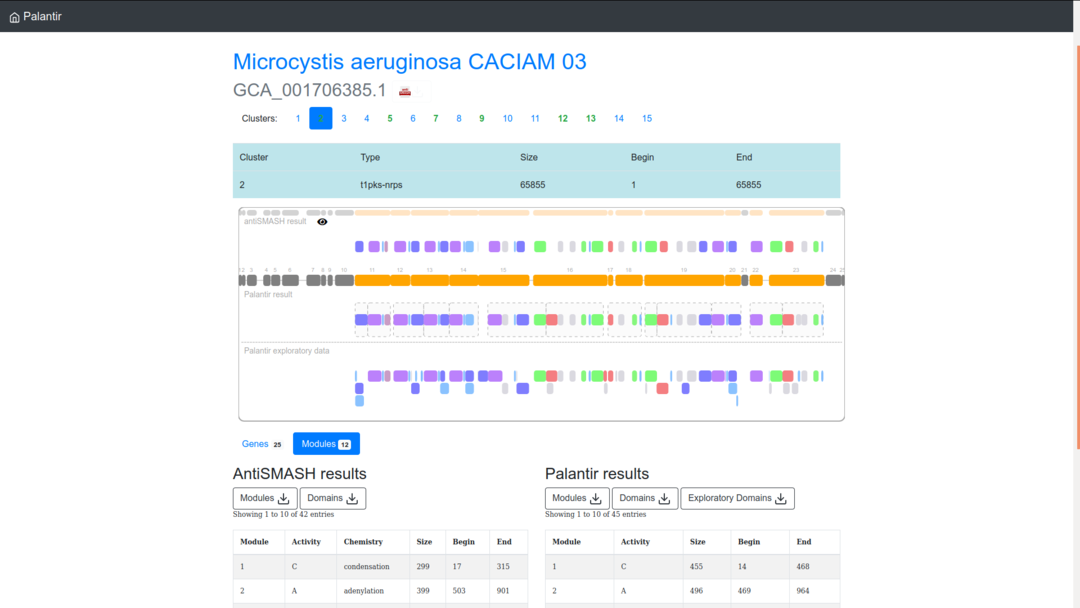
Once connected to your gene cluster of interest, appear an interface with these component (from top to bottom):
- (1) a summary line of the gene cluster numberswith in green those with a modular structure (i.e., NRPS and PKS) for which Palantir brings a new annotation;
- (2) a rectangle with gene cluster properties;
- (3) an interactive visualization of the gene cluster with its subdivisions in genes, modules and domains;
- (4) the visualization of antiSMASH domains and modules;
- (5) global gene visualization effective for the three drawing modes;
- (6) the visualization of Palantir annotation of domains and modules;
- (7) the visualization of the Exploratory annotation version of Palantir;
- (8) CSV tables to download of antiSMASH modules and domains;
- (9) CSV tables to download of Palantir modules, domains and Exploratory domains.
While Palantir functionalities improve coordinate delimitation of core NRPS/PKS domains and fill domains that fall in the exceptions of antiSMASH detection rules, the Exploratory annotation show every single domain which has a match with an E-value greater than 10e-3.
his latter annotation has the advantage to free the user from any pre-existing domain composition or architecture, and lets her/him devise her/his own view of the gene cluster based on experience, which can be useful for peculiar NRPS/PKS enzymes. However, the user needs to approach the Exploratory annotation cautiously, as false positive domains are more likely to occur. To this end, we advise examining the domain details (E-value, bit score) to assess how strong is a given prediction and if the domain has any biological meaning in its position. For example, we support the idea that when a gap seems obvious in the enzyme machinery (e.g., an NRPS with [C-gap-PCP]-[C-A-PCP]) and the missing domain is retrieved with a low evalue at the right position, this domain potentially plays a role in modular synthesis.
Palantir annotations (basic and Exploratory ones) are easy to access and use as they are available to download with data at different scales of the gene cluster under the form of tables in CSV format (Genes, Modules and Domains).
See CSV table content:
-
Genes:
- unique id;
- rank (in the cluster);
- gene name;
- the detail of the domains encoded in the gene;
- gene size in amino acids;
- protein coordinates;
- begin coordinate;
- end coordinate.
-
Modules:
- unique id;
- rank (in the cluster);
- module size in amino acids;
- protein coordinates;
- begin coordinate;
- end coordinate.
-
Domains (Palantir results): TODO
Other functionalities provided by Palantir (SQL database, JSON and PDF report generation) are only available with the stand-alone Perl package version as these are useful when applied to hundreds to thousands of antiSMASH reports (which does not seem either possible to generate with the online version of antiSMASH).